5. DIAGNÓSTICO
Deve-se suspeitar de leucodistrofia metacromática em pacientes com declínio neurológico progressivo e alterações sugestivas da doença na ressonância magnética. O diagnóstico pode ser desafiador em muitos casos, como na forma infantil tardia, pois a ressonância magnética do cérebro pode ser inicialmente normal e os primeiros sintomas (hiporreflexia e atraso no desenvolvimento) são relativamente inespecíficos.
Para investigação, são realizados testes genéticos e bioquímicos, incluindo:- Atividade da enzima arilsulfatase A em glóbulos brancos (leucócitos do sangue) ou fibroblastos (células encontradas em tecido conjuntivo, em biópsia de pele);- Medição de sulfatídeos na urina;- Teste genético molecular para variantes patogênicas nos genes ARSA e PSAP;
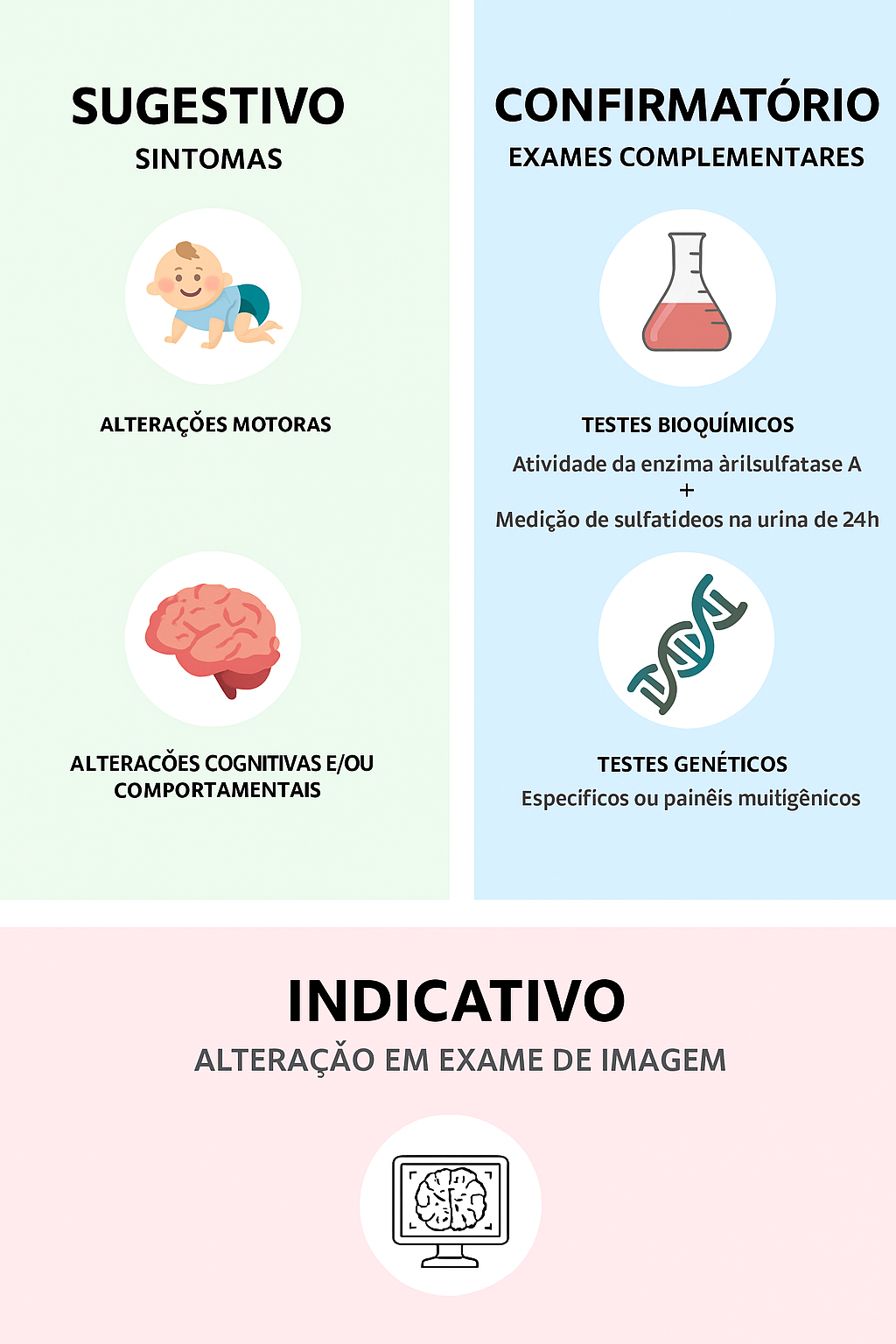
Figura 4. Critérios clínicos e laboratoriais para diagnóstico da leucodistrofia metacromática.
Tanto o ensaio enzimático quanto a medição de sulfatídeos urinários são partes essenciais do diagnóstico bioquímico, complementando o teste genético. Em um paciente com disfunção neurológica progressiva e/ou leucodistrofia, o diagnóstico é estabelecido quando observam-se alterações nos três exames.
A disponibilidade de triagem neonatal (teste do pezinho) para leucodistrofia metacromática é limitada e ainda não está disponível em todos os países.
Diagnósticos diferenciais:
- Doença de Krabbe: doença de depósito lisossomal de herança autossômica recessiva, causada pela atividade deficiente da enzima galactocerebrosidase. Clinicamente, apresenta-se com irritabilidade, hipertonia, hiperestesia e parada psicomotora.
- Adrenoleucodistrofia ligada ao X: Afeta principalmente meninos, é uma leucodistrofia que se apresenta com insuficiência adrenal, problemas neurocognitivos (baixo QI) e neurocomportamentais (comportamento semelhante ao TDAH), disartria, disgrafia, déficits na visão, audição.
- Doença de Canavan: É uma leucodistrofia de herança autossômica recessiva infantil caracterizada por deficiência intelectual, irritabilidade, macrocefalia, disfagia, hipotonia precoce, espasticidade tardia, ataxia, convulsões e atrofia óptica. Tem maior frequência na população judaica ashkenazi.
- Distúrbios da biogênese peroxissomal: Leucodistrofias causadas por mutações em um dos 13 genes PEX diferentes, que codificam as proteínas do peroxissomo, sendo PEX1 o mais comum, levando a peroxissomos disfuncionais. Os sintomas clínicos podem incluir deficiência intelectual, dismorfias craniofaciais, retinopatia (por exemplo, glaucoma, retinite pigmentosa), cegueira, surdez neurossensorial, hipotonia, hepatomegalia, coagulopatia, displasias renais, icterícia difusa, disfagia, condrodisplasia punctata e convulsões.
- Doença do corpo poliglucosano: Uma doença de herança autossômica recessiva de início adulto caracterizada por bexiga neurogênica progressiva, dificuldades de marcha (por exemplo, espasticidade e fraqueza) devido ao envolvimento misto dos neurônios motores superiores e inferiores, perda sensorial predominantemente nas extremidades inferiores distais e algumas dificuldades cognitivas (frequentemente disfunção executiva).
- Fucosidose: causada pela mutação FUCA1, caracterizada por fácies infiltrada, infecções oportunistas recorrentes, distonia generalizada, espasticidade, disostose múltipla, angioqueratoma difuso e visceromegalia..
- Esquizofrenia de início na infância: tem algumas características neurológicas e deve ser diferenciada da leucodistrofia metacromática. A esquizofrenia se apresenta com delírios, alucinações, fala desorganizada e comportamento grosseiramente desorganizado ou catatônico.
- Pseudodeficiência: Em alguns casos, indivíduos com atividade de arilsulfatase A muito baixa não desenvolvem sintomas de leucodistrofia metacromática. Essa condição é chamada de pseudo deficiência de arilsulfatase. Por este motivo é fundamental a realização da dosagem dos sulfatídeos na urina na etapa de investigação diagnóstica.